 |
第六章
基因的连锁与交换和真核生物的基因作图
|
| 第一节 基因连锁的发现生物的性别决定 | |
| 第二节 基因重组和染色体交换的作用 | |
| 第三节 交换和重组值 | |
| 第四节 基因定位和遗传作图 | |
| 第五节 真菌的遗传分析 | |
| 第六节 人类染色体作图 |
|
第六章
基因的连锁与交换和真核生物的基因作图
|
| 第一节 基因连锁的发现生物的性别决定 | |
| 第二节 基因重组和染色体交换的作用 | |
| 第三节 交换和重组值 | |
| 第四节 基因定位和遗传作图 | |
| 第五节 真菌的遗传分析 | |
| 第六节 人类染色体作图 |
第六章 基因的连锁与交换和真核生物的基因作图
基本概念:
在减数分裂时同源染色体之间的交换导致了遗传重组的发生。一个交叉(chiasm)就是交换的位点。交换是同源染色体通过联会、断裂、重接在相关位置彼此交换。
遗传重组的证据是在减数分裂中发现交换时细胞学标记发生重组,遗传标记也发生重组。
交换是一个交互事件,发生在减数分裂前期Ⅰ染色体形成二价体的阶段。
重组是交换的结果,但交换并不一定引起重组,如两标记间发生偶数次交换,标记并发生重组。所以一般交换的交换值要大于重组值。在距离较近时,双交换发生的可能性较小,故重组值近似等于交换值。在有双交换发生时,两端标记之间的距离近似等于重组值加二倍双交换值,重组频率近似于两基因间的交换频率。当基因间距离增加时,多次交换的发生可能使重组频率产生低估以至影响图距比实际的距离要小。所以要计算双交换的频率来加以校正。
两个染色体之间发生一次交叉可以阻止相邻位置第二次交叉的发生,这个现象称为交叉干涉。
孟德尔定律表明一对等位基因在遗传时相互分离,两对以上的等位基因则自由组合。遗传的染色体学说则说明基因在染色体上。基因有很多,成千上万,而染色体只有几条到几十条,最多不过几百条。例如人类有23对染色体,而基因数目为35000个。每条染色体上必然有很多基因。基因在染色体上是如何排列的呢?同一条染色体上的基因之间在遗传时又是如何相互作用的呢?
遗传学重大发现往往由一个"例外"现象而引起:
Morgan由例外的白眼雄果蝇发现了伴性遗传,他的学生Bridges由白眼雄果蝇中的初级例外和次级例外发现了染色体不分离现象,并直接证明了遗传的染色体学说。
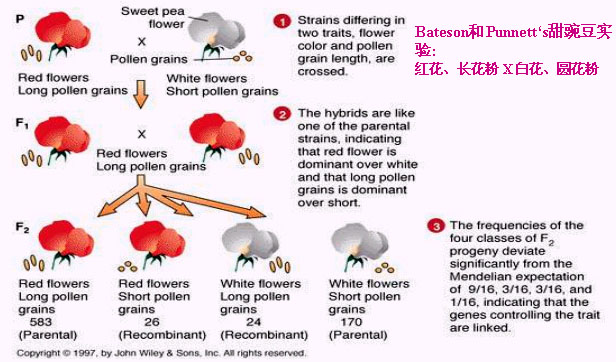
连锁现象的发现则是由两对基因杂交试验的F2代性状分离比不符合孟德尔比率9:3:3:1的例外而引起的,第一个发现非自由组合现象的是W.Bateson
和R.C.Punnet,他们于1906年在研究香豌豆的花色及花粉粒的形状的遗传时,发现这两对性状在遗传时不能自由组合。
一.相引和相斥
1905年在遗传学发展的第一个高潮时期,贝特森(Bateson,W)和庞尼特(Punet,RC)研究了香豌豆(Lathyrus
doratus)两对性状的遗传。他们选择的性状一对是花的颜色,有紫色和红色,紫色(P)对红色(p)是显性;另一对是花粉的形状,有长形(L)和圆形(l)两种,长形对圆形是显性。杂交的结果F1代都开紫色花,长形花粉,证明紫色对红色是显性,长形对圆形是显性的。在F2代中开紫花的305株,开红花的76株,带有长形花粉的是305株,圆形花粉的为76株。这个结果花的颜色和花粉形态的分离比各自符合3:1,表现都是由单基因控制的,不符合孟德尔的两对因子的分离比9:3:3:1之比。庞尼特又扩大样本重做实验,其结果和前次的相近,仍不符合孟德尔定律,亲组合比理论数多,重组合比理论数少得多,对于这一有趣的现象,庞尼特提出了新的假设,认为似乎两对基因在杂交子代中的组合并不是随机的,而是原来属于同一亲本的两个基因更倾向于进入同一配子中,这叫做相引(coupling),原来属于不同亲本的两个基因(P和l,L和p)之间形成配子时相互排斥,称为相斥(repulsion)。为了证明这个假设的正确,他们又将紫花圆形花粉和红花长形花粉的亲本杂交,结果F1代和第一次杂交相同都是紫花长形花粉,F2代有四种表型,结果也不符合孟德尔的9:3:3:1之比,虽然差异不像前次那样显著,但仍然是亲组合比理论数多。也就是说F2代性状的分离比受到亲本性状的影响;生殖细胞形成时亲组合的基因相引,重组合的基因排斥。

二.连锁法则的建立
1910年摩尔根和他的学生布里吉斯(Bridges CB)研究了两对基因的遗传,发现了连锁和互换,建立了遗传学的第三个基本定律――连锁法则。摩尔根发现果蝇的红眼和紫眼(purple)、长翅和残翅(vestigial)两对性状都是非伴性遗传,各自的遗传都符合孟德尔法则。他们将红眼长翅的果蝇和紫眼残翅的果蝇进行杂交,F1代为红眼长翅,然后将F1和双隐性的亲本进行测交,所得到的测交后代按孟德尔法则应有四种表型,分离比为1:1:1:1,但实际得到的结果确有相引和相斥的现象,亲组合多于理论数,重组合少于理论数。同贝特森等一样。为了进一步验证,他们又改变了组合重新实验,结果相似,仍是亲组合多于理论数,重组合少于理论数。
但他们的解释却不相同,摩尔根等认为第一种组合Pr+、Vg+两个基因位于同一条染色体上,Pr、Vg也位于另一亲本的相应染色体上,两个亲本都是纯合体,杂交后F1代的这一对染色体分别携带着红眼长翅和紫眼残翅基因。
减数分裂时有的细胞在Pr+、Vg+两个基因之间发生了染色体的交换重组,产生了重组型配子Pr+Vg和PrVg+,这种配子的比例比亲组合型的配子少,所以通过测交多得到的后代不是1:1:1:1,而是亲组合的表型红眼长翅和紫眼残翅为多,重组合的后代红眼残翅、紫眼长翅为少;另一杂交组合也是同样的道理。
他们的假设归纳起来主要有三个论点:1.相引就是两个基因位于同一条染色体上,相斥反之。其实按现代的概念,相引就是顺式(cis),相斥就是反式(trans)。2.同源染色体在减数分裂时发生交换(crossing-over)重组(1910年);3.1911年摩尔根又补充了一点:位置相近的因子相互连锁。连锁和交换的重组称为染色体内重组(intrachromosomal
recombination),因染色体自由组合而产生的重组称为染色体间重组(interchromosomal
recombination)。
染色体内重组是交换的结果,但交换不一定导致重组。因为我们是依据标记基因来判断是否重组的,若在两个标记基因之间发生奇数次交换,结果必然导致重组,但若发生偶数次交换,标记基因并不重组。
不能进行自由组合的基因群称为连锁群(Linkage group),排列在同一条染色体上及其同源染色体上的基因属于同一连锁群。一种二倍体生物的连锁群数应和其染色体对数相等。在少数的生物中如雄果蝇和雌家蚕都不发生重组,人们称之为完全连锁。1922年英国的霍尔丹(Haldane
JBS)提出:凡是较少发生交换的个体必定是异配性别个体,此称为霍尔丹定律。为什么在雄果蝇和雌家蚕中不会产生重组呢?根据细胞学的观点,发现在减数分裂中没有交叉,也看不到联会复合体。那么在减数分裂中性染色体如何配对又如何精确分离呢?这一机制尚待进一步研究。
根据交换发生的随机性,我们可以说距离越长,发生交换的机会越多,因此交换值的大小就可以用来表示基因间距离的长短。但我们无法直接测定交换率,只有通过标记基因的重组来估计交换的频率。
一.玉米实验
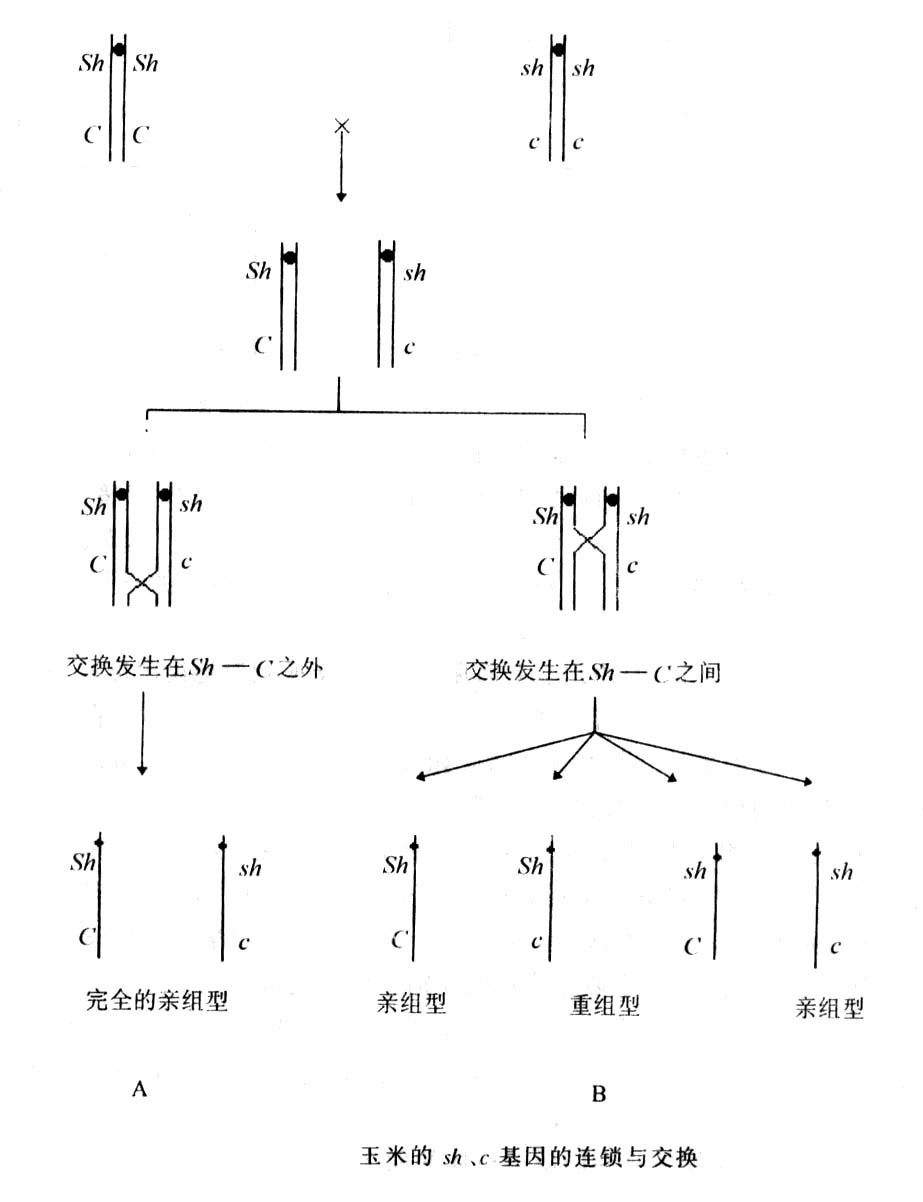
摩尔根的学说是根据实验的结果,假设在减数分裂时由于染色体的交换导致了遗传重组,虽然这一学说已被广泛接受,但毕竟是个假说,还必须进一步通过实验加以证实。1930年前都未能获得证实重组和染色体交换之间关系的有力证据。直到1931年美国著名的遗传学家麦克林托克(McClintock)指导了她的女博士生克莱顿(Creighton,B)以玉米为材料进行了一项有趣的实验,为染色体交换导致遗传重组提供了第一个有力的证据。
她们已经研究了玉米的第二个最小的染色体,即9号染色体上带有色素基因C和糯质基因wx,在其短臂上(靠近C)带有一个明显的纽结(knob)。在长臂端(靠近Wx)有一条来自第8号染色体的附加片段,正常的染色体是没有纽结和易位片段的,因此纽结和附加片段就成为一种细胞学标记。她们选用了一个杂合品系(图),其中一条染色体带有有色(C)和非糯(Wx)基因,两端有标记,而另一条染色体是正常的,两端不带有标记。这条染色体上带有的是无色基因(c)和糯质基因(wx),他们通过杂交后比较亲本型后代和重组后代的染色体,发现亲本型的后代都保持了亲本的染色体排列,而有的重组型后代的染色体也发生了重组。这样她们把遗传学和染色体内重组的细胞学证据联系起来。但"交叉即是染色体交换"这一观点直到1978年才获得证据。
摩尔根的果蝇实验已经表明重组是同源染色体发生交换的结果,两个连锁基因之间的距离决定了它们的重组值。那么我们能否用实验的方法来确定真核生物染色体上不同基因的位置呢?答案是肯定的,这种方法我们称为遗传作图(genetic mapping)。
一.三点测交
前面已经介绍基因位于染色体上,而且位置相近的基因是相互连锁的,不难想象基因在染色体上是直线排列的,但如何证明这个问题呢?摩尔根的学生斯特蒂文特(Sturtevant)想出了一个巧妙的办法,那就是三点测交(three-point
test cross)。从几何知识上知道,要证明a、b、c三点共线,可以通过三点之间的距离加以证明,当ab+bc=ac时三点在一条直线上。斯特蒂文特借鉴了这一思路,选择了六个性连锁的基因进行果蝇杂交实验,为了很好的说明问题现在我们以棘眼ec(echinus
eyes)、翅无横脉cv(cross veinless)和截翅ct(cut,or snipped wing edges)三个基因为例,杂交及结果如下。
一.链孢霉(Neurospora crassa)的生活史
链孢霉有两种繁殖方式,一种是无性繁殖,当其孢子n或菌丝落在营养物上。孢子萌发,菌丝生长形成菌丝体n。另一种繁殖方式是有性繁殖,两个亲本必须是不同的交配型(mating
type)A和a,各自的分生孢子会散落在不同交配型子囊果(perithecium,子实体)的受精丝上,进入子实体,进行核融合,形成2n核(A/a)。二倍体时期十分短暂,很快进行减数分裂,最后再经过一次有丝分裂,在子囊果中产生8个单倍体的子囊孢子,子囊孢子成熟后又可萌发长成新的菌丝体。
链孢霉后代多,样本大,统计分析的误差小,生活周期短,在短时间内可获得结果。链孢霉易培养和操作,可利用选择培养基筛选各种突变型;链孢霉又属于真核生物。可作为真核的研究模型,因此是很好的遗传学实验材料。子囊孢子是单倍体,不存在显隐性的问题,表型和基因型一致;孢子按顺序排列在子囊中,我们称其为顺序四分子(ordered
tetrad),经过分析易于确定是否是重组类型及基因的转变,其着丝粒本身也可看作一个位点来研究,因此在四分体时期进行遗传分析是很方便的,这种方法称为四分子分析(terard
analysis)。
二.着丝粒作图
链孢霉的野生型又称为原养型(prototroph),在基本培养基上就能生长,子囊孢子成熟时为黑色。有一类突变型是不能合成某些物质,称为营养缺陷型(auxotroph),在基本培养基上不能生长。如赖氨酸缺陷型是自己不能合成赖氨酸,因此培养时必须在基本培养基上加入适量的赖氨酸,此种缺陷型才能生长。缺陷型比野生型的子囊孢子成熟得慢,所以镜检时突变型的子囊孢子是白色的,和黑色的野生型子囊孢子很容易区分。
人类染色体作图也就是基因定位,随着分子生物学的飞速发展,定位的方法也越来越先进。到1998年为止人类基因图工作组(HGM)公布在常染色体上已确定480个位点,在X染色体上已确定了139个位点。
定位的方法有体细胞杂交,家系连锁分析,原位杂交,剂量效应,染色体畸变,限制性酶的精确分析等,其中前三者用得较多。
一.体细胞杂交法
人类的染色体和小鼠的染色体无论是数目还是形态都不相同,通过细胞融合可以得到人和小鼠的杂种细胞,这种细胞不稳定,在有丝分裂的过程中总是要排斥人类的染色体,最终仍随机地留下几条人类的染色体,形成不同的细胞系或"克隆",这才稳定
下来,通过Giemsa分带或其他分析方法,人们能很容易地鉴别出各细胞系中留下的人类染色体是第几号染色体,这就给基因定位提供了前提。我们可通过测定酶的活性来确定那些小鼠不含有而人类却具有的酶的基因作,以及小鼠为突变型,,人类具有其野生型等位基因的酶的位点。例如肽酶C(peptidase
C)是小鼠中没有的,而人类是可以产生此酶。若在杂种细胞系中除去小鼠的全套染色体组外还留有一条人类1号染色体,同时又测得肽酶C的活性,那就可以推测,肽酶C的基因位于人类的1号染色体上。再如胸苷激酶(TK)是小鼠和人类都有的酶,那么我们可以选择TK-小鼠的细胞和人类的细胞融合,用HAT(含有次黄嘌呤H,氨基嘌呤A和胸苷T)的培养基来选择杂种细胞,只有具有TK酶活性的杂种细胞才能存活,那么这个酶的基因一定位于杂种细胞中留有的人类染色体上。但杂种细胞留有的人类染色体常常不止一条,这样就要通过多个相关的杂种细胞系来加以比较分析(图表),从图表中我们不难推断TK位于人类的第17号染色体上。确定一个基因要多个相关克隆并非易事,因为留下的染色体是随机的,所以获得的细胞系不一定是我们所需要的,为此人们采用了克隆嵌板法(clone
panel method)来筛选就要方便得多,虽构建克隆嵌板并不容易,但一经建立便一劳永逸,事半功倍。
二.果蝇实验
在克莱顿和麦克林托克的结果发表没几周,斯特恩(Stern C)又发表了果蝇实验的证据,和玉米实验有异曲同工之妙。斯特恩选择带有异形同源染色体的雌果蝇为材料。此果蝇的一条X染色体上带有两个突变基因,一个是Car(Carnation)基因,是隐性突变,纯合体为粉红眼;另一个是B(Bareye)基因,表型为棒眼,为显性突变。这条X染色体缺失了一个片段;另一条X染色体的相应等位基因都是野生型的,但染色体带有Y染色体的易位片段,这样两条不同的X染色体都有了细胞学的标记,可供镜检分辨。雄性果蝇的X染色体上带有car和非棒眼基因,雄性是完全连锁的。在杂交后代中,不仅可以看到表型(眼色和眼形)的重组,同时在显微镜下还可以观察到带有标记染色体的重组。这两种重组又完全一致,从而证实了基因重组是交换的结果。
由于有时虽发生了交换(如双交换),但没有导致重组,因此重组值并完全等于交换值,而是重组值≤小于(或等于)交换值。重组值或重组率P ≈约为d(两个标记基因间距离的估计数)。RF=重组合/(亲组合+重组合)×100%,单位是图距单位m.u.(map
unit)或厘摩(centimorgan,cM)。重组值和实际图距的关系反应了重组值和交换值之间的关系。
霍尔丹的图谱函数是RF=1/2(1-e-2d),RF是重组频率,d是图谱距离,e是自然对数的底(e=2.71828)。在此函数中,假设没有干涉,这只是对较大的染色体图距是有效的。
从图中可以看出按霍尔丹函数作出的曲线,(1)在图距10以内曲线近似直线方程,斜率接近于1,重组率几乎等于交换率,它可以直接被看作是图距。(2)当图距大于10
时,此重组率总是小于交换率,不能直接被看作为图距,必须加双交换的值予以校正。(3)重组率最大值不可能超过50%,无论染色体上两点距离如何大,一次交换只可能发生在同源染色体的两条染色单体之间,另外两条单体并未发生交换,即使是每个初级性母细胞都如此,重组率也只有50%,其实当重组率达到50%时已是染色体间的自由组合了。

三点测交结果总结 |
|||||
表型 |
实得数 |
比例 |
重组发生在 |
||
ec-cv间 |
cv-ct间 |
ec-ct间 |
|||
ec + ct |
2125 |
||||
+ cv + |
2207 |
81.5% |
|||
ec cv + |
273 |
||||
+ + ct |
265 |
10.1% |
√ |
√ |
|
ec + + |
217 |
||||
+ cv ct |
223 |
8.3% |
√ |
||
+ + + |
5 |
||||
| ec cv ct | 3 |
0.1% | √ |
√ |
|
总计 |
5318 |
1 |
10.2% |
8.4% |
18.4% |
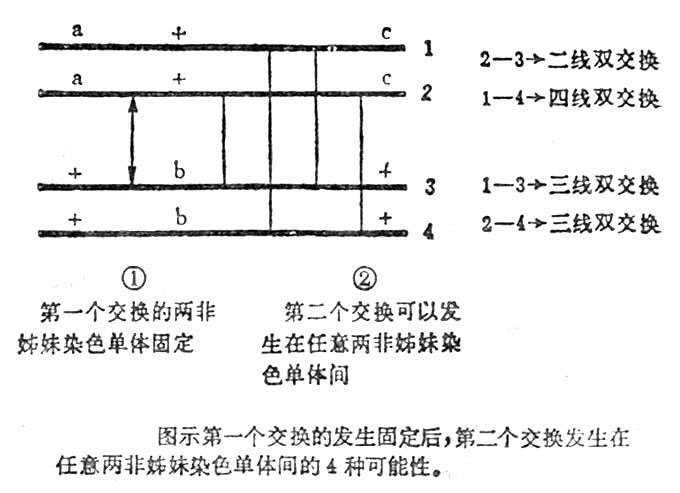
三.染色单体干涉
在减数分裂的前期,二价体有4条染色单体,交换并不限于二条非姐妹染色单体之间,而有各种不同的交换形式。为了便于说明,我们把4条染色单体分别标记上序号。当2-3之间已发生了一次交换的基础上,再发生2-3二线双交换;1-4四线双交换;1-3三线双交换。它们的机会是均等的。一般说来第一次交换仅对于2-3二线双交换有干涉,对其他类型的双交换没有干涉。

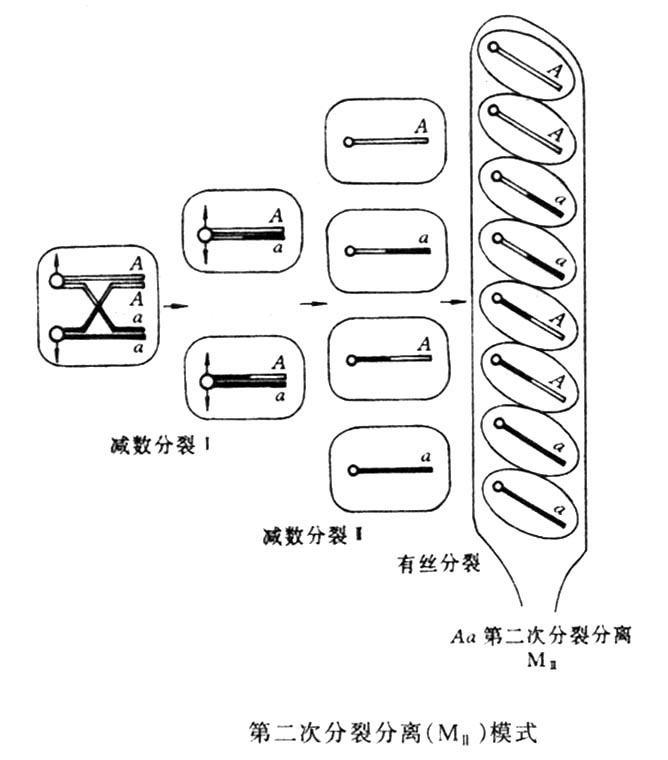
3.着丝粒作图(centromere mapping)
就是利用分裂分离来确定是否重组,再来计算标记基因到着丝粒之间的图距。计算公式为:重组率=(交换型子囊数/总子囊数)×1/2×100% 。这一公式和真核生物的重组率的计算原理相同,都是计算重组型占后代总数的比,本公式为什么乘以1/2呢?因为我们统计的单位不是子囊孢子,而是子囊。一个子囊中含有8个子囊孢子,它们来自四条染色单体,即使是重组型的子囊里面含的四条染色单体也只有两条发生了交换,还有两条未发生交换,所以必须乘以1/2。
三.链孢霉的连锁作图
链孢霉中有两种缺陷型,一种是nic(nicotine),不能合成烟酸;另一种是ad(adenine),不能合成腺嘌呤,培养时必须在培养基上分别加入烟酸和腺嘌呤。将这两个品系杂交,杂交后代中得到7种不同的子囊类型。PD为亲二型(parental
ditype),意为表型像亲本,共有+ad和nic+良种类型;NPD为非亲二型,意为表型都不像亲本,有++和nic ad两种类型;T为四型(tetratyne),意为有四种基因型++,nicad,+ad,nic+。
事先我们并不知道nic和ad基因是连锁的,通过四分体分析我们不仅能够确定这两个基因是否连锁,而且如果连锁的话我们还能确定它们在染色体上的排列顺序,计算出nic和ad与着丝粒之间的图距以及nic和ad之间的距离。

首先我们来看第一种子囊型,从nic和ad两个基因的组合(横向排列)来看,他它们是+ad和nic+两种类型,和两个亲本完全相同,故标以亲本二型(PD),这组亲组合的子囊数为808,占了整个后代子囊数(1000)的80.8%,即大部分为亲组合,少部分为重组合。由此乐意判断nic和擦的两个标记是连锁在同一条染色体上。我们再纵向来看第一纵行,为++nicnic,这种排列方式是符合第一次分裂分离(MⅠ),nic和着丝粒之间未发生交换重组。第二纵行为ad
ad + +也属于第一次分裂分离的排列方式,也表明ad与着丝粒之间未发生交换重组。
第二种子囊型nic和ad两个基因的组合方式(横向排列)也只有两种类型,++和nic ad,和亲本都不相同,故称为非亲二型(NPD)。从纵向来看,第一行的排列方式为++nicad,第二行为++adad,都属于MⅠ,即两个标记nic、擦的与着丝粒之间都未发生交换重组。既然第一类和第二类子囊都未发生重组,那么为什么它们两个标记基因的组合(横向排列)又完全不同呢?这是由于在nic座位和ad座位之间发生了1、4四线双交换的结果,图表,使得ad
与着丝粒之间仍然为第一次分离分离,实际上是发生了双交换,只是着丝粒与ad座位未发生重组。
第三类子囊的两个标记有四种不同的组合方式,成为四型(T),其中第一纵行为++nicnic,属于第二次分裂分离(MⅠ),表明nic 与着丝粒之间未发生交换重组。而第二行就不同了,排列为+ad+ad
,属于第二次分裂分离(MⅡ)⑻表明在着丝粒和ad之间曾发生过一次交换(1,4二线单交换),才能从第一类亲本的类型变为第三类子囊型。
第四类子囊也属于四型(T0,但和第三类子囊不同,其第一纵行的排列为+nic+nic,属第二次分裂分离MⅡ,第二纵行为adad++属第一次分裂分离,也就是说nic和ad
之间也发生了一次交换,但对于ad和着丝粒之间来说是发生3,4双线双交换,所以虽然发生了交换,但ad标记未发生重组。
第五类子囊两个标记的组合只有两种类型,+ad和nic+,和亲本的组合相同,所以称为PD,但第一纵行和第二纵行都属于MⅡ,即ad,nic与着丝粒之间都发生了一次交换,使这两个作为都发生了重组,实际上是在nic和着丝粒之间发生了2,3二线单交换,使nic和着丝粒发生了重组,而ad和nic是连锁的。因此也同时发生了重组。
第六类子囊型中,两个标记的组合也只有两种++和nicad,和亲本的都不同,故属于NPD,第一纵行和第二纵行的排列都是属于MⅡ,即nic和ad两个座位与着丝粒之间都发生了重组,这种类型的产生较为复杂,从两个标记的组合和纵向的排列来看,是在第四类子囊型交换重组的基础上增加了一次1,4四线双交换(图),这次双交换发生在nic和ad之间。
第七类子囊两个标记有四种组合,即为T,两列纵向排列分别为+nic+ic和+adad+,都属于MⅡ,即两个标记和着丝粒之间都发生了交换。根据两个标记的组合及纵向排列方式不难推测是在第五类子囊型交换重组的基础上产生了1,3三线双交换,这次交换应发生在ad与着丝粒之间。
根据着丝粒作图公式我们可以计算各标记与着丝粒之间的图距。着丝粒与nic座位之间的图距可通过各种子囊型中重组型的子囊数来计算,根据图表只有第4,5,6,7型子囊和着丝粒之间才存在MⅡ,即发生交换重组,所以RF(着丝粒-nic)=(4、5、6、7)/1000×1/2×100%=5.05%;同理RF(着丝粒-ad)=(3、5、6、7)/1000×1/2×100%=9.30%
。
计算的结果告诉我们nic和着丝粒之间的距离为5.05图距单位(m.u.)。ad 和着丝粒之间的距离为9.03图距单位。目前知道了nic和ad是在同一条染色体上以及它们和着丝粒的距离,但还不知道具体的顺序排列。因根据目前的信息有两种可能:1.nic和ad分别位于着丝粒的两侧;2.nic和ad位于着丝粒的同侧。究竟属于哪一种情况呢?假设两个位点在着丝粒的两侧,那么nic和着丝粒之间的重组与ad和着丝粒之间的重组是各自独立的,也就是说nic和着丝粒之间发生一次重组不会影响到ad和着丝粒之间的关系;相反若两个座位都在着丝粒的同侧,一旦nic和着丝粒之间发生了一次交换,若不存在双交换的话,势必使ad和着丝粒也产生重组。从前面的资料来看,nic和着丝粒之间产生重组的子囊为4、5、6、7,共101个子囊,其中5、6、7型子囊共96个同时也发生了ad和着丝粒之间的重组,表明基本是符合第二种情况,那么为什么有五次不同步发生重组呢?从图表不难看出,不同步的仅为第四种子囊型,两个座位分别为MⅡ、MⅠ,共5个子囊,从重组的图中很清楚地看到是由于在nic和ad之间发生了一次双交换,结果使ad和着丝粒之间未能重组。
因ad和着丝粒图距为9.03m.u.,nic和着丝粒的图距为5.05m.u.,几乎相差一倍,而重组是一个随机的过程,因此ad与着丝粒之间发生重组的交换点可能在着丝粒-nic的区域中也可以发生在nic-ad的范围内。若是前者,则nic和ad同时发生重组。ad和着丝粒重组的子囊(MⅡ)共186个,其中nic未同步重组的为(MⅠMⅡ)为90个子囊,近似于ad重组子囊的一半,这和两个座位的距离之比是吻合的。
下面我们再来计算nic和ad之间的距离。根据计算重组值的原则:重组值=重组合/(亲组合+重组合)×100% ,那么ad和nic之间重组依据是什么呢?图表已表明两个标记的亲组合只有两种类型+ad和nic+,不同于这两种类型的子囊都四重组型。因此用下面公式可计算nic和ad之间的图距:RF=(NDD+1/2T)/总子囊数={(2)+(6)+1/2[(3)+(4)+(7)]}/1000=5.2%,NPD的子囊(2)和(6)型4条染色单体都是重组型的,即整个子囊都是重组型,故无需再乘以0.5,而T子囊中只有一半的染色单体为重组型,故需乘以0.5。
从公式计算出nic和ad之间的图距为5.20m.u.,而从图,我们用ad-着丝粒图距减去nic-着丝粒图距也应等于nic-ad图距,即9.03-5.05=3.98(m.u.),比用公式计算的图距要小,或者说着丝粒ad图距应为5.05+5.20=10.25m.u.,这样就大于前面计算的结果(9.30)。这是为什么呢?这是由于双交换的存在使着丝粒-ad之间的重组值低估了,从图表中可以看出低估的原因。
从图表可以看出着丝粒-nic + nic-ad=202+208=410≠着丝粒-ad(372),这是因为遗漏了38条重组型单体。现在的计算方法不是以子囊数而是染色单体数,所以分母应用子囊数乘4,即低估的重组值为38/4×1000=0.95,9.30+0.95=10.25m.u.。

二、 原位分子杂交
细胞融合的方法只能将某个基因定位于某条染色体上,但却无法确定其具体的位置。用玻片原位分子杂交就可以将已知的基因定位于染色体的一定区域。方法是这样的。首先要制备较好的人类中期染色体片,经变性处理使其DNA双链打开,然后将同位素标记或带有荧光标记的基因探针(probe)和玻片上的DNA进行分子杂交,经放射自显影或在荧光显微镜下观察摄影,就乐意确定该基因在染色体的具体位置。如人类的胰岛素基因就是用这种方法
三、 家系分析法
家系分析法是通过分析家系中连锁基因的重组来确定同一条染色体上基因的排列顺序以及两个基因之间的遗传距离。应当说比原位杂交的结果更为精确,但由于人类的家系相对比较小,世代很长,同时不能像动物那样进行杂交实验,因此这种方法的使用受到了很大的局限,一般较适合X连锁基因的定位,直到20世纪60年代后期人们一直都采用这种方法定位。
其他的一些定位方法如细胞学方法和基因剂量效应法是应用于一些特殊的情况。如968年约翰霍普金斯大学的学生Donahue在做染色体实验时发现自己的1号染色体缢痕区增长,通过对他自己家族染色体的观察,进一步发现这一异常是遗传的,并与特殊血型(Fy)具有平行性,因此将血型基因定位于1号染色体上,这是首次定位常染色体上的基因,从而建立了细胞学定位法。1973年发现第二条染色体短臂缺失(2P-)的患者红细胞酸性磷酸酶(ACP-1)活性明显降低,于是将此酶基因定位于第2条染色体上,这就是利用剂量效应的方法。
Dulbecco于1986年首次提出了"人类基因组工程",以期能确定人类及几种模型生物整个基因组的序列,得到了很多著名科学家的支持,这一浩大的国际协作研究计划终于在990年正式开始实施。据估计人类的基因组约含30亿个碱基对,有10万个基因分散在基因组中,其中5000个已被编目,1900个已被定位,600个已被克隆分离出来。一旦这项宏伟蓝图得以实现完成,那么基因定位将会迎刃而解。)